NUVAXOVID JN.1
Laboratoire : Sanofi Winthrop Industrie
Vaccin adapté au variant JN.1 du SARS-CoV-2.
Calendrier vaccinal 2026 : le vaccin Nuvaxovid peut être utilisé, au même titre que les vaccins à ARNm, dès lors qu’il présente le même niveau d’adaptation aux souches circulantes les plus récentes.
Description
▼ Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Nuvaxovid JN.1 dispersion injectable
Vaccin contre la covid 19 (recombinant, avec adjuvant)
Forme et Présentation
Dispersion injectable (injection).
Nuvaxovid JN.1 dispersion injectable en seringue préremplie.
La dispersion est incolore à légèrement jaune, limpide à légèrement opalescente (pH 7,2).
Nature et contenu de l'emballage extérieur
Flacon unidose
- 0,5 mL de dispersion dans un flacon (verre de type I) avec un bouchon (caoutchouc de bromobutyle) et une bague en aluminium avec une capsule amovible en plastique bleu.
Chaque flacon contient une dose de 0,5 mL.
Présentation : 1 flacon unidose ou 10 flacons unidoses
Seringue préremplie
- 0,5 mL de dispersion dans une seringue préremplie (verre de type I) avec un bouchon-piston et un capuchon de protection, sans aiguille ou avec une aiguille séparée fournie dans le même emballage.
Chaque seringue préremplie contient une dose de 0,5 mL.
Présentation :
10 seringues préremplies
1 seringue préremplie
1 seringue préremplie avec aiguille séparée
Composition
Un flacon unidose (0,5 mL) contient :
1. Antigènes
- 5 microgrammes de la protéine* Spike de SARS-CoV-2 (Omicron JN.1) et un adjuvant Matrix-M.
Contenu de l’adjuvant Matrix-M dans chaque dose de 0,5 mL : fraction A (42,5 microgrammes) et fraction C (7,5 microgrammes) d’extrait de Quillaja Saponaria Molina.
2. Excipients
- Hydrogénophosphate disodique heptahydraté
- Dihydrogénophosphate sodique monohydraté
- Chlorure de sodium
- Polysorbate 80
- Hydroxyde de sodium (pour l’ajustement du pH)
- Acide chlorhydrique (pour l’ajustement du pH)
- Eau pour préparations injectables
Adjuvant (Matrix-M)
- Cholestérol
- Phosphatidylcholine (dont all-rac-α-tocophérol)
- Dihydrogénophosphate de potassium
- Chlorure de potassium
- Hydrogénophosphate disodique dihydraté
- Chlorure de sodium
- Eau pour préparations injectables
Indications
Nuvaxovid JN.1 est indiqué pour l’immunisation active afin de prévenir la covid 19 causée par le virus SARS-CoV-2 chez les personnes âgées de 12 ans et plus.
L’utilisation de ce vaccin doit être conforme aux recommandations officielles.
Posologie
Nuvaxovid JN.1 est administré par voie intramusculaire en dose unique (0,5 mL) chez des personnes âgées de 12 ans et plus indépendamment du statut vaccinal antérieur.
Chez les personnes ayant déjà été vaccinées par un vaccin contre la covid 19, Nuvaxovid JN.1 doit être administré au moins 3 mois après la dose la plus récente d’un vaccin contre la covid 19.
Personnes immunodéprimées
Des doses supplémentaires peuvent être administrées à des personnes sévèrement immunodéprimées, conformément aux recommandations nationales, voir rubrique "Mises en garde et précautions d'emploi".
Population pédiatrique
La sécurité et l’efficacité de Nuvaxovid JN.1 chez les enfants âgés de moins de 12 ans n’ont pas encore été établies. Aucune donnée n’est disponible.
Population âgée
Aucun ajustement posologique n’est nécessaire chez les personnes âgées de ≥ 65 ans.
Mode d'administration
Nuvaxovid JN.1 doit être exclusivement administré par injection intramusculaire, de préférence dans le muscle deltoïde du haut du bras.
Ne pas injecter le vaccin par voie intravasculaire, sous-cutanée ou intradermique.
Le vaccin ne doit pas être mélangé à d’autres vaccins ou produits médicamenteux dans la même seringue.
Pour les précautions à prendre avant l’administration du vaccin, voir rubrique "Mises en garde et précautions d'emploi".
Pour les instructions concernant la manipulation et l’élimination du vaccin, voir rubrique "Manipulation".
Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique"Composition".
Mises en garde et précautions d'emploi
Traçabilité
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Recommandations générales
Hypersensibilité et anaphylaxie
Des cas d’anaphylaxie ont été signalés avec Nuvaxovid. Il convient de toujours disposer d’un traitement médical approprié et de surveiller la personne vaccinée en cas de survenue d’une réaction anaphylactique suite à l’administration du vaccin.
Une surveillance étroite pendant au moins 15 minutes est recommandée après la vaccination. Une dose supplémentaire de vaccin ne doit pas être administrée aux personnes ayant présenté une anaphylaxie après une dose antérieure de Nuvaxovid.
Myocardite et péricardite
Il existe un risque accru de myocardite et de péricardite après la vaccination par Nuvaxovid. Ces pathologies peuvent se développer quelques jours seulement après la vaccination et sont principalement survenues dans les 14 jours, voir rubrique "Effets indésirables".
Les données disponibles suggèrent que l’évolution des cas de myocardite et de péricardite après vaccination n’est pas différente des myocardites ou péricardites en général. Les professionnels de santé doivent être attentifs aux signes et symptômes de myocardite et de péricardite. Les personnes vaccinées (notamment les parents et les aidants) doivent être informées qu’elles doivent consulter immédiatement un médecin si elles présentent des symptômes évocateurs de
myocardite ou de péricardite tels qu’une douleur thoracique (aiguë et persistante), un essoufflement ou des palpitations après la vaccination.
Les professionnels de santé doivent consulter les recommandations et/ou des spécialistes pour diagnostiquer et traiter ces affections.
Réactions liées à l’anxiété
Des réactions liées à l’anxiété, notamment des réactions vasovagales (syncope), une hyperventilation ou des réactions liées au stress peuvent survenir en association à la vaccination, reflétant une réaction psychogène à l’injection via une aiguille. Il est important que des précautions soient prises afin
d’éviter toute blessure en cas d’évanouissement.
Affections concomitantes
La vaccination doit être reportée chez les personnes atteintes d’une affection fébrile aiguë sévère ou d’une infection aiguë. La présence d’une infection mineure et/ou d’une fièvre peu élevée ne doit pas retarder la vaccination.
Thrombocytopénie et troubles de la coagulation
Comme avec les autres injections intramusculaires, le vaccin doit être administré avec prudence chez les personnes recevant un traitement anticoagulant ou chez celles présentant une thrombocytopénie ou un autre trouble de la coagulation (tel que l’hémophilie), car un saignement ou une ecchymose peut survenir après une administration intramusculaire chez ces personnes.
Personnes immunodéprimées
L’efficacité, la sécurité d’emploi et l’immunogénicité du vaccin ont été évaluées chez un nombre limité de personnes immunodéprimées. L’efficacité de Nuvaxovid peut être réduite chez les personnes immunodéprimées.
Durée de la protection
La durée de la protection conférée par le vaccin n’est pas établie et est toujours en cours d’évaluation dans les essais cliniques en cours.
Limites de l’efficacité du vaccin
Les personnes pourraient ne pas être entièrement protégées jusqu’à 7 jours après l’administration de leur dose de vaccin. Comme pour tout vaccin, il est possible que les personnes vaccinées par Nuvaxovid ne soient pas toutes protégées.
Excipients
Sodium
Ce vaccin contient moins de 1 mmol (23 mg) de sodium par dose, c’est à dire qu’il est essentiellement « sans sodium ».
Potassium
Ce vaccin contient moins de 1 mmol (39 mg) de potassium par dose, c’est à dire qu’il est essentiellement « sans potassium »
Interactions
L’administration concomitante de Nuvaxovid (original, souche Wuhan) et de vaccins inactivés contre la grippe a été évaluée chez un nombre limité de participants dans une sous-étude d’essai clinique exploratoire, voir rubrique "Effets indésirables" et rubrique "Pharmacodynamie".
La réponse en anticorps de liaison dirigés contre le SARS-CoV-2 était plus faible lorsque Nuvaxovid était administré concomitamment avec un vaccin inactivé contre la grippe. La signification clinique de cette observation n’est pas connue.
L’administration concomitante de Nuvaxovid JN.1 et d’autres vaccins n’a pas été étudiée.
Fertilité
Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects
sur la toxicité de la reproduction, voir rubrique "Autres informations / Données de sécurité préclinique".
Grossesse
Il existe des données limitées sur l’utilisation de Nuvaxovid chez la femme enceinte. Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects sur la gestation, le développement embryo-fœtal, la mise-bas ou le développement postnatal, voir rubrique "Autres informations / Données de sécurité préclinique".
L’utilisation de Nuvaxovid JN.1 chez la femme enceinte doit être envisagée seulement si les bénéfices potentiels l’emportent sur les risques potentiels pour la mère et le fœtus.
Allaitement
On ne sait pas si Nuvaxovid JN.1 est excrété dans le lait maternel.
Aucun effet chez les nouveau-nés ou nourrissons allaités n’est attendu dans la mesure où l’exposition systémique à Nuvaxovid chez la femme qui allaite est négligeable.
Effets indésirables
Nuvaxovid (original, souche Wuhan)
Résumé du profil de sécurité d’emploi après la primo-vaccination
Participants âgés de 18 ans et plus
Les effets indésirables les plus fréquents après l’administration d’une dose de Nuvaxovid dans le cadre de la primo-vaccination étaient une sensibilité au site d’injection (75 %), une douleur au site d’injection (62 %), de la fatigue (53 %), des myalgies (51 %), des maux de tête (50 %), un malaise (41 %), des arthralgies (24 %) et des nausées ou vomissements (14 %). Les effets indésirables étaient généralement de sévérité légère à modérée avec une durée médiane après la vaccination inférieure ou égale à 2 jours pour les événements locaux et inférieure ou égale à 1 jour pour les événements systémiques.
Globalement, l’incidence de certains effets indésirables était plus élevée chez les participants plus jeunes : chez les adultes âgés de 18 à moins de 65 ans que chez ceux âgés de 65 ans et plus. Les effets indésirables locaux et systémiques ont été rapportés plus fréquemment après la dose 2 qu’après la dose 1.
Après l’administration concomitante avec le vaccin contre la grippe saisonnière, des fréquences plus élevées d’effets indésirables locaux au site d’injection de Nuvaxovid (70,1 % après la dose 1 et 85,0 % après la dose 2) et d’effets indésirables systémiques (60,1 % après la dose 1 et 69,7 % après la dose 2) ont été observées.
Adolescents âgés de 12 à 17 ans
La sécurité d’emploi de Nuvaxovid a été évaluée chez des adolescents dans le cadre d’une analyse intermédiaire de la partie d’expansion pédiatrique d’une étude de phase 3, multicentrique, randomisée, avec observateur en aveugle, contrôlée contre placebo, en cours (étude 2019nCoV-301). Les données sur la sécurité d’emploi ont été recueillies aux États-Unis chez 2 232 participants âgés de 12 à 17 ans, avec ou sans signe d’infection antérieure par le SARS-CoV-2, ayant reçu au moins une dose de Nuvaxovid (n = 1 487) ou de placebo (n = 745). Les données démographiques étaient similaires entre les participants ayant reçu Nuvaxovid et ceux ayant reçu le placebo.
Les effets indésirables les plus fréquents étaient une sensibilité au site d’injection (71 %), une douleur au site d’injection (67 %), des maux de tête (63 %), des myalgies (57 %), de la fatigue (54 %), un malaise (43 %), des nausées ou vomissements (23 %), des arthralgies (19 %) et de la pyrexie (17 %). Comparativement aux adultes, la fièvre a été observée plus fréquemment chez les adolescents de 12 à 17 ans, et a été avérée comme très fréquente après la deuxième dose chez les adolescents. Les effets indésirables étaient généralement de sévérité légère à modérée avec une durée médiane après la vaccination inférieure ou égale à 2 jours pour les événements locaux et inférieure ou égale à 1 jour pour les événements systémiques.
Résumé du profil de sécurité d’emploi après la dose de rappel
Participants âgés de 18 ans et plus
Les effets indésirables les plus fréquents observés après avoir reçu une dose de rappel de Nuvaxovid après la primo-vaccination à deux doses étaient une sensibilité au site d’injection (73 %), une douleur au site d’injection (61 %), de la fatigue (53 %), des myalgies (52 %), des maux de tête (46 %), un
malaise (41 %) et des arthralgies (26 %).
Adolescents âgés de 12 à 17 ans
La sécurité d’emploi d’une dose de rappel de Nuvaxovid a été évaluée dans une analyse intermédiaire d’une étude de phase 3 en cours (étude 2019nCoV-301). Au total, 1 499 participants ont reçu une dose de rappel environ 9 mois après la dose 2 du schéma de primo-vaccination. Les effets indésirables sollicités ont été évalués dans les 7 jours suivant une dose de rappel dans un sous-groupe de 220 participants ayant reçu la dose de rappel (ensemble d’analyse de la sécurité d’emploi du rappel ad hoc), parmi lesquels 190 ont rempli le journal électronique.
Comparativement aux adultes, les effets indésirables sollicités se sont produits à des fréquences plus élevées et à un grade supérieur chez les adolescents. Les effets indésirables sollicités les plus fréquents étaient une sensibilité au site d’injection (72 %), des maux de tête (68 %), de la fatigue (66 %), une douleur au site d’injection (64 %), des myalgies (62 %), un malaise (47 %) et des nausées ou vomissements (26 %) avec une durée médiane de 1 à 2 jours après la vaccination. Aucun nouvel élément en termes de sécurité d’emploi n’a été observé chez les participants du moment de l’administration de la dose de rappel jusqu’à 28 jours après l’administration
Nuvaxovid JN.1 (Nuvaxovid adapté au variant Omicron)
La sécurité d’emploi de Nuvaxovid JN.1 est déduite des données de sécurité du vaccin Nuvaxovid (original, souche Wuhan) et des données de sécurité du vaccin adapté à Omicron BA.5.
Une dose de rappel des vaccins Nuvaxovid monovalent Omicron BA.5 et bivalent original/Omicron BA.5 a été évaluée dans une étude de phase 3 en cours menée chez des participants âgés de 18 ans et plus (2019nCoV-311 partie 2). Dans cette étude, 251 participants ont reçu une dose de rappel de Nuvaxovid (original, souche Wuhan), 254 participants ont reçu une dose de rappel du vaccin monovalent Omicron BA.5, et 259 participants ont reçu une dose de rappel de Nuvaxovid bivalent original/Omicron BA.5. La durée médiane du suivi à partir du rappel initial était de 48 jours au moment de la date limite de recueil des données, le 31 mai 2023.
Le profil de sécurité global pour les doses de rappel du vaccin monovalent Nuvaxovid Omicron BA.5 était similaire à celui observé après la dose de rappel de Nuvaxovid (original, souche Wuhan). Les effets indésirables les plus fréquents étaient une sensibilité au site d’injection (> 50 %), une douleur au site d’injection (> 30 %), de la fatigue (> 30 %), des maux de tête (> 20 %), des myalgies (> 20 %), et un malaise (> 10 %). Aucun nouvel effet indésirable n’a été identifié pour les doses de rappel du vaccin monovalent Nuvaxovid Omicron BA.5. Dans la partie 2 de l’étude 2019nCoV-311, la fréquence des événements de réactogénicité locaux et systémiques était plus élevée chez les femmes que chez les hommes, pour toutes les versions du vaccin réévaluées.
Liste des effets indésirables sous forme de tableau
Sauf mention contraire, les catégories de fréquence sont basées sur la sécurité du Nuvaxovid évaluée dans 5 essais cliniques portant sur un total de 30 070 participants âgés de 18 ans et plus ayant reçu au moins une dose de la primo-vaccination à deux doses de Nuvaxovid (la durée médiane du suivi était de 84 jours après la deuxième dose) et dans un essai clinique au cours duquel 13 354 participants ont reçu une dose de rappel du vaccin au moins 6 mois après la primo-vaccination à deux doses (délai médian de 11 mois entre la fin de la primo-vaccination et la dose de rappel).
Les effets indésirables observés au cours des études cliniques sont indiqués ci-dessous selon les catégories de fréquence suivantes :
- Très fréquent (≥ 1/10)
- Fréquent (≥ 1/100, < 1/10)
- Peu fréquent (≥ 1/1 000, < 1/100)
- Rare (≥ 1/10 000, < 1/1 000)
- Très rare (< 1/10 000)
- Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Affections hématologiques et du système lymphatique
- Peu fréquent : lymphadénopathie.
Affections du système immunitaire
- Fréquence indéterminée : anaphylaxie.
Affections du système nerveux
- Très fréquent : maux de tête
- Fréquence indéterminée : paresthésie, hypoesthésie.
Affections cardiaques
- Fréquence indéterminée : myocardite, péricardite.
Affections vasculaires
- Peu fréquent : hypertension d.
Affections gastro-intestinales
- Très fréquent : nausées ou vomissements a.
Affections de la peau et du tissu sous-cutané
- Peu fréquent : éruption cutanée, érythème, prurit, urticaire.
Affections musculo-squelettiques et systémiques
- Très fréquent : myalgie a, arthralgie a.
Troubles généraux et anomalies au site d’administration
- Très fréquent : sensibilité au site d’injection a, douleur au site d’injection a, fatigue a, malaisea a,b.
- Fréquent : rougeur au site d’injection a,c, gonflement au site d’injection a, pyrexiee, douleurs des extrémités.
- Peu fréquent : prurit au site d’injection, frissons.
- Rare : Chaleur au site d'injection.
b Ce terme comprenait également des effets rapportés de syndrome pseudo-grippal.
c Ce terme comprenait à la fois la rougeur au site d’injection et l’érythème au site d’injection (fréquent).
d L’hypertension n’a pas été rapportée chez les adolescents âgés de 12 à 17 ans de l’étude clinique.
e Comparativement aux adultes, la pyrexie a été observée plus fréquemment chez les adolescents de 12 à 17 ans, et a été avérée comme très fréquente après la deuxième dose chez les adolescents.
Description de certains effets indésirables
Dans l’ensemble des essais cliniques, une hypertension a été observée à une incidence accrue au cours des 3 jours suivant la vaccination avec Nuvaxovid (n=46, 1,0 %) par rapport au placebo (n=22, 0,6 %) chez les adultes âgés.
Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
Nuvaxovid JN.1 n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines. Toutefois, certains des effets indiqués à la rubrique 4.8 peuvent temporairement altérer l’aptitude à conduire des véhicules ou à utiliser des machines.
Surdosage
Aucun cas de surdosage n’a été rapporté. En cas de surdosage, il est recommandé de contrôler les fonctions vitales et d’envisager un traitement symptomatique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration – voir Annexe V et incluent le numéro de lot s’il est disponible.
Pharmacodynamie
Classe pharmacothérapeutique : vaccin protéique sous-unitaire.
Code ATC : J07BN04.
Mécanisme d’action
Nuvaxovid JN.1 est composé de la protéine Spike (S) recombinante de SARS-CoV-2 Omicron JN.1 purifiée de pleine longueur stabilisée dans la conformation de préfusion. L’ajout de l’adjuvant Matrix-M à base de saponine facilite l’activation des cellules du système immunitaire inné, ce qui améliore l’ampleur de la réponse immunitaire spécifique à la protéine S. Les deux composants du vaccin induisent des réponses immunitaires en lymphocytes B et T contre la protéine S, notamment des anticorps neutralisants pouvant contribuer à la protection contre la covid 19.
Nuvaxovid JN.1 (Nuvaxovid adapté au variant Omicron)
L’efficacité de Nuvaxovid JN.1 est déduite des données d’efficacité du vaccin Nuvaxovid (original, souche Wuhan) et des données d’immunogénicité du vaccin adapté à la souche Omicron BA.5.
Dans l’étude 2019nCoV-311 partie 2, un total de 694 participants âgés de 18 ans et plus qui ont fait l’objet d’une évaluation de l’immunogénicité et avaient précédemment reçu 3 doses ou plus du vaccin contre la covid 19 de Pfizer-BioNTech ou du vaccin contre la covid 19 de Moderna, ont reçu une dose de rappel avec Nuvaxovid (original, souche Wuhan), ou le vaccin Nuvaxovid monovalent Omicron BA.5 ou le vaccin Nuvaxovid bivalent original/Omicron BA.5. Les doses de rappel ont été administrées avec un intervalle médian de 11 à 13 mois après la dernière vaccination respectivement. Les ratios de moyenne géométrique des titres (GMR) et les taux de séroconversion ont été évalués 1 mois après la vaccination.
L’objectif principal de l’étude était de démontrer la supériorité des titre d’anticorps neutralisants de pseudovirus (ID50) et la non-infériorité des taux de séroconversion de la réponse immunitaire contre Omicron BA.5 induite par une dose du vaccin Nuvaxovid bivalent original/Omicron BA.5 par rapport à la réponse obtenue avec une dose de Nuvaxovid (original, souche Wuhan) et d’évaluer la non-infériorité des titres d’anticorpsID50 contre la souche originale du SAR-CoV-2 pour le vaccin Nuvaxovid bivalent original/Omicron BA.5 par rapport à Nuvaxovid (original, souche Wuhan).
La supériorité de l’ID50 contre-Omicron BA.5 du vaccin Nuvaxovid bivalent original/Omicron BA.5 par rapport à Nuvaxovid (original, souche Wuhan) a été démontrée, la borne inférieure de l’intervalle
de confiance (IC) à 95 % bilatéral pour le GMR étant > 1. La non-infériorité de l’ID50 contre la souche originale du vaccin Nuvaxovid bivalent original/Omicron BA.5 par rapport à Nuvaxovid (original, souche Wuhan) a été satisfaite, la limite inférieure de l’IC à 95 % bilatéral pour le GMR étant > 0,67. La non-infériorité du taux de séroréponse contre le variant Omicron BA.5 du vaccin Nuvaxovid bivalent original/Omicron BA.5 par rapport à Nuvaxovid (original, souche Wuhan) a été satisfaite, la limite inférieure de l’IC à 95 % bilatéral pour la différence des pourcentages de participants ayant présenté une séroréponse étant > -5 %. Pour de plus amples détails, consulter le Tableau 1.
Les analyses exploratoires de l’immunogénicité comprenaient une évaluation du ratio des GMT ID50 et des différences en termes de taux de séroréponse pour le vaccin monovalent Nuvaxovid Omicron BA.5 par rapport à Nuvaxovid (original, souche Wuhan). Le ratio des GMT suite au rappel avec le vaccin Nuvaxovid monovalent Omicron BA.5 par rapport à la dose de rappel par Nuvaxovid (original, souche Wuhan) était de 2,5 (IC à 95 % bilatéral : 2,10 ; 2,94). La différence des taux de séroconversion entre la dose de rappel avec le vaccin Nuvaxovid monovalent Omicron BA.5 et la dose de rappel avec Nuvaxovid (original, souche Wuhan), était de 33,2 % (IC à 95 % bilatéral : 25,4 ; 40,7). Bien que n’ayant pas été formellement évaluées, ces réponses auraient répondu aux trois critères de
réussite de l’étude.
Tableau 1 : Titres (ID50) d’anticorps neutralisants contre les pseudovirus Omicron BA.5 et Wuhan et taux de séroréponse suite au rappel avec le vaccin Nuvaxovid monovalent BA.5, Nuvaxovid (original, souche Wuhan) et Nuvaxovid bivalent Original/Omicron BA.5 – Sous-groupe de test de neutralisation de pseudovirus PP ; étude 2019nCoV-311 Partie 2


1 La valeur de référence était définie comme la dernière évaluation non manquante avant le rappel vaccinal.
2 L’IC à 95 % pour le GMT et la GMFR a été calculé sur la base de la distribution t de Student des valeurs transformées en log puis remises à l’échelle d’origine en vue de la présentation.
3 Une ANCOVA avec groupe de vaccin et tranche d’âge (18-54 ans, ≥ 55 ans) comme effets fixes, et la valeur de référence (Jour 0) comme covariable, a été réalisée en incluant tous les groupes des vaccins afin d’estimer le GMT pour tous les groupes des vaccins. Chaque comparaison par pair incluait les données issues de deux groupes seulement afin d’estimer le GMTR ajusté entre les deux groupes des vaccins. La différence moyenne entre groupes des vaccins et les limites de l’IC correspondant étaient ensuite exponentialisées afin d’obtenir le ratio des GMT ID50 ainsi que les IC à 95 % correspondants.
4 Le TSR était défini comme le pourcentage de participants lors de chaque visite post-vaccination présentant un titre augmenté ≥ 4 fois du niveau ID50 par rapport à l’inclusion si la valeur de référence est supérieure ou égale à la LIdQ ou ≥ 4 fois la LIdQ si la valeur de référence est inférieure à la LIdQ puis calculé sur la base de n2 comme dénominateur.
5 L’IC à 95 % pour le TSR a été calculé à l’aide de la méthode de Clopper-Pearson.
6 L’IC à 95 % pour la différence de TSR a été calculé à l’aide de la méthode Miettinen et Nurminen.
Nuvaxovid (original, souche Wuhan)
Mécanisme d’action
Nuvaxovid JN.1 est composé de la protéine Spike (S) recombinante de SARS-CoV-2 Omicron JN.1 purifiée de pleine longueur stabilisée dans la conformation de préfusion. L’ajout de l’adjuvant Matrix-M à base de saponine facilite l’activation des cellules du système immunitaire inné, ce qui améliore l’ampleur de la réponse immunitaire spécifique à la protéine S. Les deux composants du vaccin induisent des réponses immunitaires en lymphocytes B et T contre la protéine S, notamment des anticorps neutralisants pouvant contribuer à la protection contre la covid 19.
Efficacité clinique
Schéma de primo-vaccination
L’efficacité clinique, la sécurité et l’immunogénicité de Nuvaxovid sont en cours d’évaluation dans deux études pivots de phase 3 contrôlées contre placebo, l’étude 1 (2019nCoV-301) menée en Amérique du Nord et l’étude 2 (2019nCoV-302) menée au Royaume-Uni et dans une étude de phase 2a/b, l’étude 3 conduite en Afrique du Sud.
Étude 1 (2019nCoV-301)
L’étude 1 était une étude de phase 3, multicentrique, randomisée, avec observateur en aveugle, contrôlée contre placebo, avec une étude principale chez les adultes menée chez des participants âgés de 18 ans et plus aux États-Unis et au Mexique et une expansion pédiatrique réalisée chez des participants âgés de 12 à 17 ans aux États-Unis.
Participants âgés de 18 ans et plus
Lors de leur inclusion dans l’étude principale chez les adultes, les participants ont été stratifiés selon leur âge (18 à 64 ans ou ≥ 65 ans) et randomisés selon un ratio 2:1 afin de recevoir Nuvaxovid ou le placebo. L’étude a exclu les participants sévèrement immunodéprimés en raison de leur état clinique ; atteints d’un cancer traité par chimiothérapie ; ayant reçu un traitement chronique par immunosuppresseur, des immunoglobulines ou des produits dérivés du sang dans les 90 jours précédents ; les femmes enceintes ou qui allaitent et les participants ayant un antécédent de covid 19 biologiquement confirmé. Des participants présentant des comorbidités stables sur le plan clinique ont été inclus, de même que des participants présentant une infection par le VIH bien contrôlée.
L’inclusion des adultes s’est terminée en février 2021. Les participants ont été suivis jusqu’à 24 mois après la deuxième dose afin d’évaluer la sécurité et l’efficacité contre la COVID-19. Après le recueil de données de sécurité suffisantes pour appuyer la demande de mise sur le marché conditionnelle, les premiers participants ayant reçu le placebo ont été invités à recevoir deux injections de Nuvaxovid à 21 jours d’intervalle et les premiers participants ayant reçu Nuvaxovid ont été invités à recevoir deux injections de placebo à 21 jours d’intervalle (« étude croisée en aveugle »). Tous les participants ont eu la possibilité de continuer à être suivis dans l’étude.
La population de l’analyse principale de l’efficacité (correspondant à la population d’analyse de l’efficacité selon le protocole [Per-Protocol Efficacy, PP-EFF]) comprenait 25 452 participants ayant reçu Nuvaxovid (n = 17 312) ou le placebo (n = 8 140), à raison de deux doses (dose 1 le Jour 0 ; dose 2 le Jour 21, médiane 21 jours [IIQ : 21 - 23], extrême 14 - 60), n’ayant pas présenté de déviation majeure au protocole et n’ayant eu aucun signe d’infection par le SARS-CoV-2 jusqu’à 7 jours après la deuxième dose.
Les données démographiques à l’inclusion étaient équilibrées entre les participants ayant reçu Nuvaxovid et ceux ayant reçu le placebo. Dans la population d’analyse PP-EFF, chez les participants ayant reçu Nuvaxovid, l’âge médian était de 47 ans (extrême : 18 à 95 ans) ; 88 % (n = 15 264) étaient âgés de 18 à 64 ans et 12 % (n = 2 048) étaient âgés de 65 ans et plus ; 48 % étaient des femmes ; 94 % vivaient aux États-Unis et 6 % au Mexique ; 76 % étaient Caucasiens, 11 % étaient Afro-américains, 6 % étaient des Indiens américains (comprenant les Amérindiens) ou autochtones d’Alaska et 4 % étaient asiatiques ; 22 % étaient hispaniques ou latino-américains. Au moins une comorbidité préexistante ou un facteur de risque lié au mode de vie associé à un risque accru de forme sévère de covid 19 était présente chez 16 493 (95 %) participants. Les comorbidités comprenaient : obésité (définie par un indice de masse corporelle [IMC] ≥ 30 kg/m2) ; maladie pulmonaire chronique ; diabète de type 2, maladie cardiovasculaire ; maladie rénale chronique ; ou virus de l’immunodéficience humaine (VIH). Les autres facteurs de risque comprenaient un âge ≥ 65 ans (avec ou sans comorbidités) ou un âge < 65 ans associé à des comorbidités et/ou des conditions de vie ou de travail impliquant une exposition fréquente au SARS-CoV-2 ou à des zones densément peuplées.
Les cas de covid 19 ont été confirmés par réaction en chaîne par polymérase (PCR) dans un laboratoire central. L’efficacité du vaccin est présentée dans le Tableau 2.
Tableau 2. Efficacité du vaccin contre la covid 19 : cas confirmés par PCR survenus à partir de 7 jours après la deuxième dose1 – Population d’analyse PP-EFF ; étude 2019nCoV-301
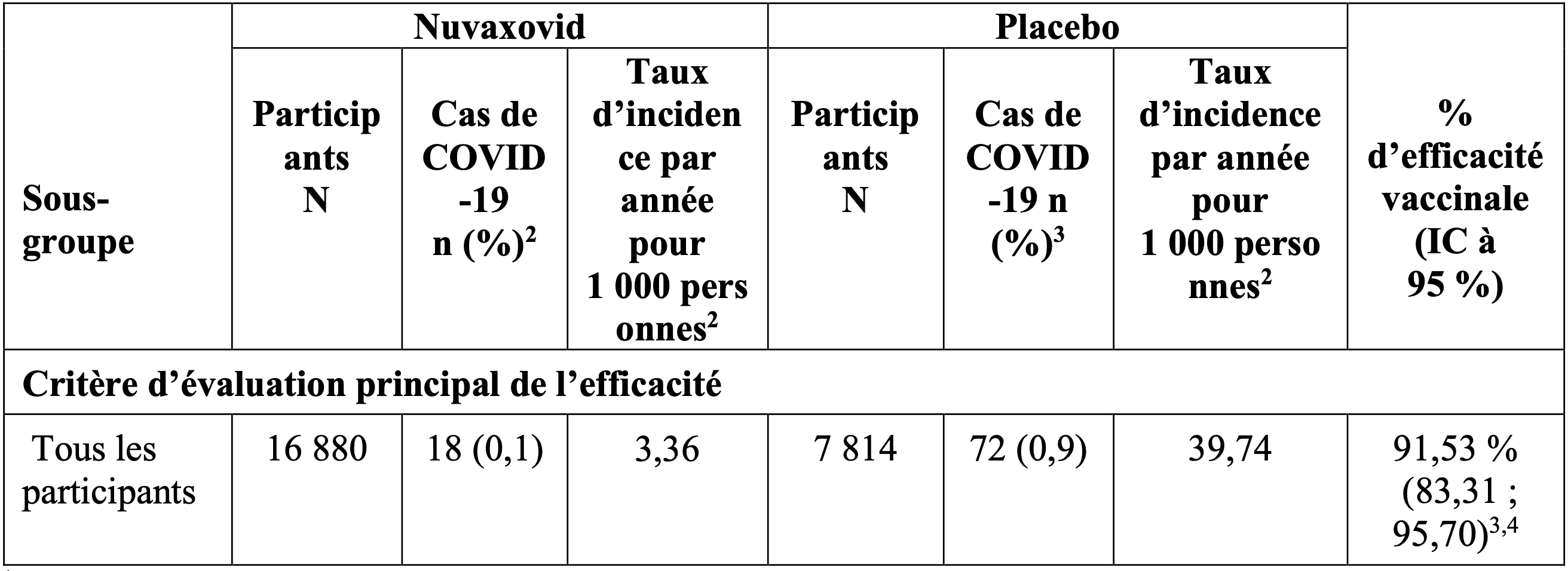
2 Taux moyen d’incidence de la maladie par an pour 1 000 personnes.
3 D’après un modèle log-linéaire du taux d’incidence d’infection covid 19 confirmée par PCR utilisant une régression de Poisson avec les strates de groupe de traitement et d’âge comme effets fixes et une variance d’erreur robuste, pour laquelle EV = 100 × (1 – risque relatif) (Zou 2004).
4 Critère d’évaluation principal de l’efficacité atteint pour la réussite avec une limite inférieure de l’intervalle de confiance (LIIC) > 30 % lors de l’analyse principale confirmatoire prévue.
L’efficacité du vaccin Nuvaxovid pour prévenir la survenue de la COVID-19 à partir de sept jours après la dose 2 était de 91,53 % (IC à 95 % : 83,31 ; 95,70). Aucun cas de forme sévère de la COVID-19 n’a été rapporté chez les 16 880 participants ayant reçu Nuvaxovid comparativement à 4 cas de forme sévère de la COVID-19 rapportés chez les7 886 participants ayant reçu le placebo dans la population d’analyse PP-EFF.
Des analyses en sous-groupes du critère d’évaluation principal de l’efficacité ont montré une efficacité vaccinale estimée similaire chez les participants de sexe masculin ou féminin, selon l’origine ethnique et chez les participants présentant des comorbidités associées à un risque élevé de covid 19 sévère. Concernant l’efficacité vaccinale globale, aucune différence significative n’a été observée chez les participants qui présentaient un risque accru de forme sévère de covid 19, notamment ceux ayantune ou plusieurs comorbidités augmentant le risque de forme sévère de covid 19 (par ex., IMC≥ 30 kg/m2, maladie pulmonaire chronique, diabète de type 2, maladie cardiovasculaire et maladierénale chronique).
Les résultats d’efficacité sont basés sur une période d’inclusions qui a eu lieu au moment où les souches classées comme variants préoccupants ou variants surveillés circulaient principalement dans les deux pays (États-Unis et Mexique) où l’étude a été menée. Les données de séquençage étaient disponibles pour 70 des 90 cas du critère d’évaluation principal (78 %). Parmi ceux-ci, 54 sur 70 (77 %) ont été identifiés comme des variants préoccupants ou des variants surveillés. Les variants préoccupants/surveillés les plus fréquemment identifiés étaient Alpha avec 52/90 cas (58 %), Bêta (2/90 cas, 2 %), Gamma (3/90 cas, 3 %), Iota avec 9/90 cas (10 %) et epsilon (19/90 cas, 21 %).
Efficacité chez les adolescents âgés de 12 à 17 ans
L’efficacité et l’immunogénicité de Nuvaxovid chez les participants adolescents âgés de 12 à 17 ans a été évaluée aux États-Unis dans la partie d’expansion pédiatrique de l’étude 2019nCoV-301 de phase 3, multicentrique, randomisée, avec observateur en aveugle, contrôlée contre placebo, en cours. Au total, 1 799 participants affectés selon un ratio 2:1 pour recevoir deux doses de Nuvaxovid (n = 1 205) ou de placebo (n = 594) par injection musculaire à 21 jours d’intervalle constituaient la population d’efficacité per protocole. Les participants avec une infection confirmée ou une infection antérieure par le SARS CoV-2 au moment de la randomisation n’étaient pas inclus dans l’analyse principale de l’efficacité.
L’inclusion des adolescents s’est achevée en juin 2021. Les participants ont été suivis jusqu’à 24 mois après la deuxième dose afin d’évaluer la sécurité d’emploi, l’efficacité et l’immunogénicité contre la covid 19. Après une période de suivi de la sécurité d’emploi de 60 jours, les premiers adolescents ayant reçu le placebo ont été invités à recevoir deux injections de Nuvaxovid à 21 jours d’intervalle et les premiers participants ayant reçu Nuvaxovid ont été invités à recevoir deux injections de placebo à 21 jours d’intervalle (« étude croisée en aveugle »). Tous les participants ont eu la possibilité de continuer à être suivis dans l’étude.
La covid 19 était définie comme un premier épisode de covid 19 léger, modéré ou sévère confirmée par PCR avec au moins l’un des symptômes prédéfinis au sein de chaque catégorie de sévérité. La covid 19 légère était définie par de la fièvre, une toux nouvellement apparue ou au moins 2 ou plus symptômes supplémentaires de covid 19.
Vingt cas de covid 19 légers symptomatiques confirmés par PCR ont été observés (Nuvaxovid, n = 6 [0,5 %] ; placebo, n = 14 [2,4 %]), ce qui conduit à une estimation ponctuelle de l’efficacité de 79,5 % (IC à 95 % : 46,8 %, 92,1 %).
Au moment de cette analyse, le variant préoccupant Delta (lignées B.1.617.2 et AY) était le variant prédominant circulant aux États-Unis et représentait tous les cas pour lesquels les données de séquence étaient disponibles (11/20, 55 %).
Immunogénicité chez les adolescents âgés de 12 à 17 ans
La réponse en anticorps neutralisant le SARS-CoV-2 14 jours après la dose 2 (Jour 35) a été analysée chez les participants adolescents séronégatifs pour la nucléoprotéine (NP) anti-SARS-CoV-2 et négatifs pour la PCR à l’inclusion. Les réponses en anticorps neutralisants ont été comparées avec celles observées chez les participants adultes séronégatifs et négatifs pour la PCR, âgés de 18 ans à 25 ans, dans l’étude principale chez les adultes (population d’immunogénicité per protocole [Per Protocol Immunogenicity, PP-IMM]), comme indiqué dans le Tableau 3. La non-infériorité exigeait que soient satisfaits les trois critères suivants : limite inférieure de l’IC bilatéral à 95 % pour le ratio de moyenne géométrique des titres (geometricmeantiters, GMT) (GMT de 12 à 17 ans/GMT de 18 à 25 ans) > 0,67 ; estimation ponctuelle du ratio de GMT ≥ 0,82 ; et la limite inférieure de l’IC bilatéral à 95 % pour la différence des taux de séroconversion (seroconversion rates, SCR) (SCR de 12 à 17 ans moins SCR de 18 à 25 ans) > -10 %. Ces critères de non-infériorité ont été atteints.
Tableau 3. Ratio ajusté de moyenne géométrique des titres d’anticorps neutralisants pour le virus SARS-CoV-2 S de type sauvage déterminés par un test de microneutralisation au Jour 35, globalement et par tranche d’âge (population d’analyse PP-IMM)
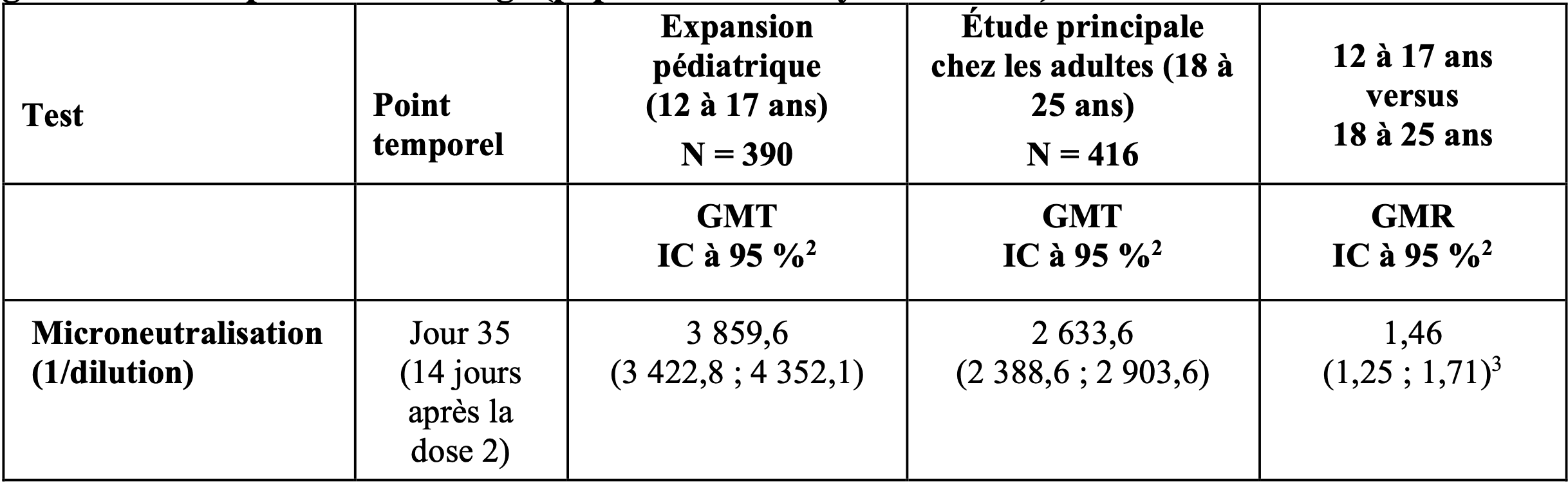
1 Le tableau inclut uniquement les participants dans le groupe de vaccin actif.
2 Une ANCOVA avec la cohorte d’âge comme effet principal et les anticorps neutralisants du test MN à l’inclusion comme covariable a été réalisée pour estimer le GMR. Les valeurs de réponse individuelle enregistrées comme étant inférieures à la LIdQ ont été définies à la moitié de la LIdQ.
3 Représente les populations (n1, n2) définies comme suit :
n1 = nombre de participants dans l’étude principale chez l’adulte (18 à 25 ans) avec un résultat des anticorps neutralisants non manquant
n2 = nombre de participants dans l’expansion pédiatrique (12 à 17 ans) avec un résultat des anticorps neutralisants nonmanquant
Étude 2 (2019nCoV-302)
L’étude 2 était une étude de phase 3, multicentrique, randomisée, avec observateur en aveugle, contrôlée contre placebo, menée chez des participants âgés de 18 à 84 ans au Royaume-Uni. Lors de l’inclusion, les participants ont été stratifiés selon leur âge (18 à 64 ans ou 65 à 84 ans) et ont été randomisés afin de recevoir Nuvaxovid ou le placebo. L’étude a exclu les participants sévèrement immunodéprimés en raison de leur état clinique ; atteints d’un cancer traité ou non par chimiothérapie ; ayant une maladie auto-immune ; ayant reçu un traitement chronique immunosuppresseur, des immunoglobulines ou des produits dérivés du sang dans les 90 jours précédents ; présentant un trouble de la coagulation ou sous anticoagulants de façon continue; ayant des antécédents de réaction allergique et/ou d’anaphylaxie ; les femmes enceintes ; ou les participants ayant un antécédent de diagnostic de covid 19 biologiquement confirmé. Des participants présentant une maladie stable sur le plan clinique, définie comme une maladie ne nécessitant pas de changement significatif de traitement ou une hospitalisation en raison de l’aggravation de la maladie au cours des 4 semaines précédant l’inclusion ont été inclus. Les participants présentant une infection connue stable par le VIH, le virus de l’hépatite C (VHC) ou le virus de l’hépatite B (VHB) n’étaient pas exclus de l’inclusion.
L’inclusion s’est achevée en novembre 2020. Les participants ont été suivis jusqu’à 12 mois après le schéma de primo-vaccination afin d’évaluer la sécurité et l’efficacité contre la covid 19.
La population de l’analyse principale de l’efficacité (PP-EFF) comprenait 13 971 participants ayant reçu Nuvaxovid (n=6 979) ou le placebo (n=6 992), à raison de deux doses (dose 1 le Jour 0 ; dose 2 après une médiane de 21 jours [IIQ : 21 - 23], extrême 16 - 45), n’ayant pas présenté de déviation majeure au protocole et n’ayant eu aucun signe d’infection par le SARS-CoV-2 jusqu’à 7 jours après la deuxième dose (Tableau 4).
Les données démographiques à l’inclusion étaient équilibrées entre les participants ayant reçu Nuvaxovid et ceux ayant reçu le placebo. Dans la population d’analyse PP-EFF, chez les participants ayant reçu Nuvaxovid, l’âge médian était de 56,0 ans (extrême : 18 à 84 ans) ; 72 % (n=5 039) étaient âgés de 18 à 64 ans et 28 % (n = 1 940) étaient âgés de 65 à 84 ans ; 49 % étaient des femmes ; 95 % étaient Caucasiens, 3 % étaient asiatiques, < 1 % avaient plusieurs origines ethniques, < 1 % étaient Afro-américains et < 1 % étaient hispaniques ou latino-américains ; 45 % présentaient au moins une comorbidité.
L’efficacité du vaccin Nuvaxovid pour prévenir l’apparition de la COVID-19 à partir de 7 jours après la dose 2 était de 87,2 % (IC à 95 % : 78,1 ; 92,5). Aucun cas de COVID-19 sévère n’a été signalé parmi les 6 979 participants ayant reçu Nuvaxovid, contre 6 cas de COVID-19 sévère parmi les 6 992 participants ayant reçu le placebo dans la population d’analyse PP-EFF.
Tableau 4. Analyse de l’efficacité vaccinale contre la covid 19 : cas confirmés par PCR survenus à partir de 7 jours après la deuxième dose – (population PP-EFF) : étude 2 (2019nCoV-302)
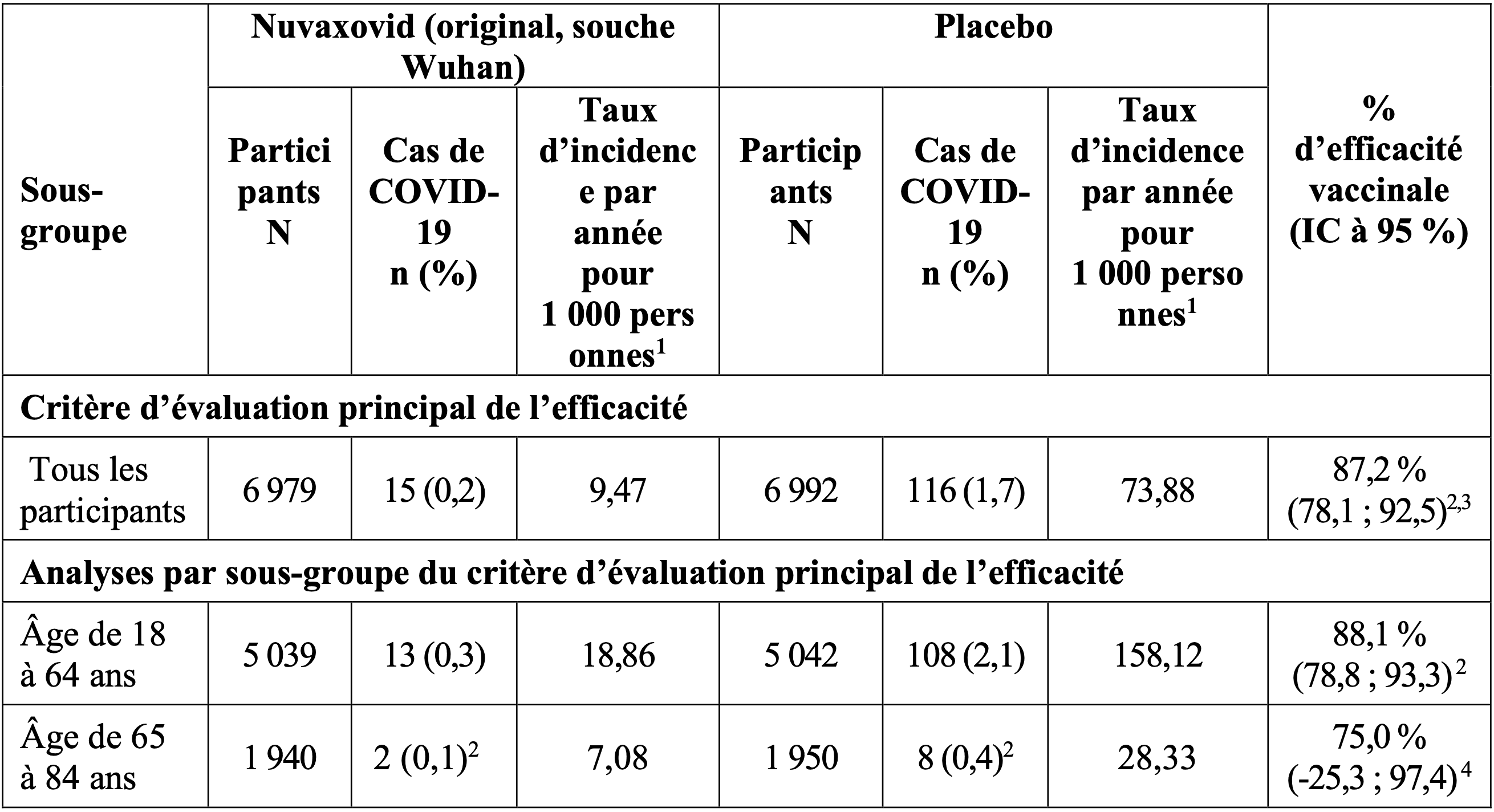
2 D’après un modèle log-linéaire de survenue utilisant une régression de Poisson modifiée avec une fonction de lien logarithmique, avec le groupe de traitement et la strate (tranche d’âge et région regroupée) comme effets fixes et variance d’erreur robuste [Zou 2004].
3 Critère d’évaluation principal de l’efficacité atteint pour la réussite avec une limite inférieure de l’intervalle de confiance (LIIC) > 30 %, l’efficacité a été confirmée lors de l’analyse intermédiaire.
4 Basé sur le modèle de Clopper-Pearson (en raison du faible nombre d’événements), les IC à 95 % ont été calculés par la méthode binomiale exacte de Clopper-Pearson ajustée selon la durée totale de surveillance.
Ces résultats sont basés sur une période d’inclusions qui a eu lieu au moment où le variant B.1.1.7 (Alpha) circulait au Royaume-Uni. L’identification du variant alpha était basée sur l’échec de la cible du gène S par PCR. Les données étaient disponibles pour 118 des 131 cas du critère d’évaluation principal (90 %). Parmi ceux-ci, 80/118 cas (68 %) ont été identifiés comme étant dus au variant Alpha, et les autres cas étaient classés en non-Alpha.
Sous-étude d’administration concomitante d’un vaccin homologué contre la grippe saisonnière
Sous-étude d’administration concomitante d’un vaccin homologué contre la grippe saisonnière Dans l’étude, 429 participants ont été vaccinés concomitamment par des vaccins inactivés contre la grippe saisonnière ; 217 participants à cette sous-étude ont reçu Nuvaxovid et 212 ont reçu le placebo. Les données démographiques à l’inclusion étaient équilibrées entre les participants ayant reçu Nuvaxovid et ceux ayant reçu le placebo. Dans la population d’analyse de l’immunogénicité selon le protocole (per-protocol immunogenicity, PP-IMM), chez les participants ayant reçu Nuvaxovid (n=190), l’âge médian était de 40 ans (intervalle : 22 à 70 ans) ; 94 % (n=178) étaient âgés de 18 à 64 ans et 6 % (n=12) étaient âgés de 65 à 84 ans ; 43 % étaient des femmes ; 86 % étaient Caucasiens, 14 % avaient plusieurs origines ethniques ou étaient issus de minorités ethniques ; et 27 % présentaient au moins un comorbidité. La co-administration n’a pas entraîné de changement de réponse immunitaire du vaccin contre la grippe, mesurée par un test d’inhibition de l’hémagglutination (IHA). Une réduction de 30 % de la réponse anticorps de Nuvaxovid a été observée, telle que mesurée par un dosage des IgG anti-protéine Spike, avec des taux de séroconversion similaires à ceux des participants qui n’avaient pas reçu concomitamment le vaccin contre la grippe, voir rubrique "Interactions" et Effets indésirables".
Étude 3 (2019nCoV-501)
L’étude 3 est une étude de phase 2a/b, multicentrique, randomisée, avec observateur en aveugle, contrôlée contre placebo, menée chez des participants séronégatifs pour le VIH, âgés de 18 à 84 ans et chez des personnes vivant avec le VIH (PVVIH) âgées de 18 à 64 ans en Afrique du Sud. Les PVVIH étaient stables sur le plan clinique (sans infections opportunistes), recevaient un traitement antirétroviral hautement actif et stable et présentaient une charge virale du VIH-1 < 1 000 copies/ mL.
L’inclusion s’est achevée en novembre 2020.
La population de l’analyse principale de l’efficacité (PP-EFF) comprenait 2 769 participants ayant reçu Nuvaxovid (n = 1 413) ou le placebo (n = 1 356), à raison de deux doses (dose 1 le Jour 0 ; dose 2 le Jour 21), n’ayant pas de déviation majeure au protocole et n’ayant aucun signe d’infection par le SARS-CoV-2 jusqu’à 7 jours après la deuxième dose.
Les données démographiques à l’inclusion étaient équilibrées entre les participants ayant reçu Nuvaxovid et ceux ayant reçu le placebo. Dans la population d’analyse PP-EFF, chez les participants ayant reçu Nuvaxovid, l’âge médian était de 28 ans (extrême : 18 à 84 ans) ; 39 % étaient des femmes ; 94 % étaient Afro-américains, 5 % étaient Caucasiens, 3 % avaient plusieurs origines ethniques, 1 % étaient asiatiques et 2 % étaient hispaniques ou latino-américains ; et 5,4 % étaient séropositifs pour le VIH.
Au total, l’analyse complète du critère d’évaluation principal de l’efficacité regroupait 168 cas de COVID-19 aux symptômes légers, modérés ou sévères, parmi tous les participants adultes, séronégatifs (pour le SARS-CoV-2) à l’inclusion (population d’analyse PP-EFF), dont 57 cas (4,0 %) avaient reçu Nuvaxovid et 111 cas (8,2 %) avaient reçu le placebo. Cette analyse a montré une efficacité du vaccin Nuvaxovid de 50,7 % (IC à 95 % : 32,8 ; 63,9).
Ces résultats sont basés sur une période d’inclusions qui a eu lieu au moment où le variant B.1.351 (bêta) circulait en Afrique du Sud.
Dose de rappel
Immunogénicité chez des participants âgés de 18 ans et plus
Étude 2019nCoV-101, partie 2
La sécurité et l’immunogénicité d’une dose de rappel de Nuvaxovid ont été évaluées dans une étude de phase 2, randomisée, avec observateur en aveugle, contrôlée contre placebo. Une dose de rappel unique Nuvaxovid a été administrée chez des participants adultes en bonne santé, âgés de 18 à 84 ans, séronégatifs pour le SARS-CoV-2 à l’inclusion (étude 2019nCoV-101, partie 2). Au total, 254 patients (ensemble d’analyse complet) ont reçu un schéma de primo-vaccination comprenant deux doses de Nuvaxovid (0,5 mL, 5 microgrammes à 3 semaines d’intervalle). Un sous-groupe de 104 participants a reçu une dose de rappel de Nuvaxovid environ 6 mois après la dose 2 du schéma de primo-vaccination. Une dose unique de rappel de Nuvaxovid a induit une multiplication par environ 84,8 des anticorps neutralisants, passant d’un GMT de 63 avant le rappel (Jour 189) à un GMT de 5 834,3 après le rappel (Jour 217), et une multiplication par environ 6,8 du pic de GMT à 855,2 (14 jours après la dose 2).
Étude 2019nCoV-501
Dans l’étude 3, étude de phase 2a/b, randomisée, avec observateur en aveugle, contrôlée contre placebo, la sécurité et l’immunogénicité d’une dose de rappel ont été évaluées chez des participants adultes non infectés par le VIH, en bonne santé, âgés de 18 à 84 ans et chez des PVVIH stables sur le plan médical, âgées de 18 à 64 ans, séronégatives pour le SARS-CoV-2 à l’inclusion. Au total, 1 169 participants (ensemble d’analyse PP-IMM) ont reçu une dose de rappel de Nuvaxovid environ 6 mois après la fin du schéma de primo-vaccination par Nuvaxovid (Jour 201). Une multiplication par environ 52,2 des anticorps neutralisants, passant d’un GMT de 69 avant le rappel (Jour 201) à un GMT de 3 603 après le rappel (Jour 236), et une multiplication par environ 5,2 du pic de GMT à 690 (14 jours après la dose 2) ont été observées.
La sécurité et l’immunogénicité des vaccins contre la COVID-19 administrés en doses de rappel après la fin d’un schéma de primo-vaccination avec un autre vaccin contre la COVID-19 autorisé ont été évaluées lors d’une étude indépendante menée au Royaume-Uni.
L’essai de phase 2 institutionnel, indépendant, multicentrique, randomisé, contrôlé (CoV-BOOST, EudraCT 2021-002175-19) a étudié l’immunogénicité d’un rappel chez des adultes âgés de 30 ans et plus, sans antécédents d’infection par le SARS-CoV-2 biologiquement confirmée. Nuvaxovid a été administré au moins 70 jours après la fin d’un schéma de primo-vaccination par ChAdOx1 nCoV-19 (Oxford-AstraZeneca) ou au moins 84 jours après la fin d’un schéma de primo-vaccination par BNT162b2 (Pfizer-BioNTech). Les titres d’anticorps neutralisants mesurés par un test avec le virus de type sauvage ont été évalués 28 jours après la dose de rappel. Dans le groupe ayant reçu une dose unique de rappel Nuvaxovid (0,5 mL), 115 participants avaient reçu un schéma de primo-vaccination en deux doses par ChAdOx1 nCoV-19 et 114 participants avaient reçu un schéma de primo-vaccination en deux doses par BNT162b2. Nuvaxovid (original, souche Wuhan) a démontré une réponse mémoire, quel que soit le vaccin utilisé pour la primo-vaccination.
Dose de rappel chez les adolescents âgés de 12 à 17 ans
L’efficacité des doses de rappel de Nuvaxovid chez les adolescents âgés de 12 à 17 ans a été extrapolée à partir de données obtenues chez les adultes ayant reçu une dose de rappel Nuvaxovid dans les études 2019nCoV-101 et 2019nCoV-501. Il a été montré que la réponse immunitaire et l’efficacité étaient comparables chez les adolescents et les adultes ayant reçu le schéma de primo-vaccination, et que la dose de rappel avait stimulé la réponse immunitaire chez les adultes.
Population âgée
Nuvaxovid a été évalué chez des individus âgés de 18 ans et plus. L’efficacité de Nuvaxovid chez les sujets âgés (≥ 65 ans) était en cohérence avec celle observée chez les sujets adultes plus jeunes (18 à 64 ans) pour le schéma de primo-vaccination.
Population pédiatrique
L’Agence européenne des médicaments a différé l’obligation de soumettre les résultats d’études réalisées avec Nuvaxovid dans un ou plusieurs sous-groupes de la population pédiatrique en prévention de la covid 19 (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Conservation
À conserver au réfrigérateur (entre 2 °C - 8 °C).
Ne pas congeler.
Conserver les flacons dans l’emballage extérieur à l’abri de la lumière.
Flacon non ouvert
9 mois entre 2 °C et 8 °C, à l’abri de la lumière.
Le vaccin Nuvaxovid non ouvert s’est avéré stable jusqu’à 12 heures à 25 °C. La conservation à 25 °C n’est pas la condition de conservation ou de transport recommandée mais elle peut guider des décisions d’utilisation en cas d’écarts temporaires de température au cours des 9 mois de conservation entre 2 °C et 8 °C.
Flacon entamé
D’un point de vue microbiologique, le vaccin doit être utilisé immédiatement après l’ouverture. Le flacon unidose doit être jeté après le retrait et l’administration d’une dose, voir rubrique "Manipulation".
Manipulation
Instructions pour la manipulation et l’administration
Ce vaccin doit être manipulé par un professionnel de santé en respectant les règles d’asepsie afin d’assurer la stérilité de chaque dose.
Préparation à l’utilisation
- Le vaccin est fourni prêt à l’emploi.
- Le vaccin non ouvert doit être conservé entre 2 °C et 8 °C et maintenu dans l’emballage extérieur à l’abri de la lumière.
- Juste avant utilisation, sortez le flacon de vaccin de son emballage du réfrigérateur.
- Jetez le flacon et tout volume résiduel après le retrait et l’administration d’une dose de 0,5 mL.
Inspection du flacon
- Faites doucement tournoyer le flacon avant et entre chaque retrait de dose. Ne le secouez pas.
- Chaque flacon contient une dispersion incolore à légèrement jaune, limpide à légèrement opalescente sans aucune particule visible.
- Inspectez visuellement le contenu du flacon pour vérifier l’absence de particules visibles et/ou de changement de couleur avant l’administration. N’administrez pas le vaccin en présence de ces signes.
Administration du vaccin
- Le flacon contient un volume excédentaire afin de s’assurer qu’une dose de 0,5 mL puisse être prélevée à partir d’un flacon unidose.
- Chaque dose de 0,5 mL est prélevée à l’aide d’une aiguille et d’une seringue stériles et est administrée par injection intramusculaire, de préférence dans le deltoïde en haut du bras.
- Ne mélangez pas le vaccin avec d’autres vaccins ou médicaments dans la même seringue.
Mise au déchet
- Jetez le flacon et tout volume résiduel après le retrait et l’administration d’une dose.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation envigueur.
Autres informations
Les données non cliniques issues des études conventionnelles de toxicologie en administration répétée, de tolérance locale et de toxicologie sur la reproduction et le développement n’ont pas révélé de risque particulier pour l’homme.
Génotoxicité et carcinogénicité
Des études de génotoxicité in vitro ont été menées avec l’adjuvant Matrix-M. Il a été démontré que l’adjuvant est non génotoxique. Aucune étude de carcinogénicité n’a été réalisée. Aucune carcinogénicité n’est anticipée.
Toxicité pour la reproduction
Une étude de toxicologie sur le développement et la reproduction a été menée chez des rats femelles ayant reçu quatre doses intramusculaires (deux avant l’accouplement ; deux au cours de la gestation) de 5 microgrammes de protéine S recombinante de SARS-CoV-2 (environ 200 fois plus que la dose humaine de 5 microgrammes sur une base ajustée selon le poids) avec 10 microgrammes d’adjuvant Matrix-M (environ 40 fois plus que la dose humaine de 50 microgrammes sur une base ajustée selon le poids). Aucun effet indésirable lié au vaccin n’a été observé sur la fertilité, la grossesse, l’allaitement ou le développement de l’embryon ou du fœtus et de la progéniture jusqu’au Jour 21 post-natal.
Pour les dernières mises à jour, voir RCP sur le site l'EMA.